
 Your Idea
Your Idea
 Research Planning
Research Planning
 Project Planning
Project Planning
 Impact
Impact
NIHR look for details within the project plan to support the compilation of information for future CE/UKCA marking and other regulatory requirements, including any work toward Quality Management System (QMS) development and reporting on clinical evaluation.
Definitions
Medical device - any instrument, apparatus, appliance, software, implant, reagent, material or other article intended by the manufacturer to be used, alone or in combination, for human beings.
In vitro diagnostic medical device - any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body.
Manufacturer - a natural or legal person who manufactures or fully refurbishes a device or has a device designed, manufactured or fully refurbished, and markets that device under its name or trademark.
Regulatory Compliance and Steps Required Towards UKCA/CE Mark Approval
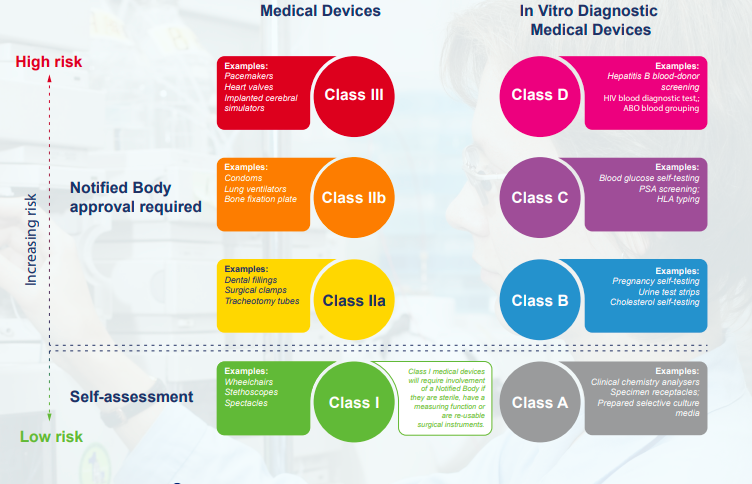
Classification - Classification is based on risk and evidence needs to be compiled throughout the project to ensure that the medical device will meet requirements for regulatory approval and will meet conformity assessment if required.
To meet conformity assessment requirements:
- Outline of general safety and performance
- Benefits must outweigh risks
- Must be supported by clinical evidence and evaluation
- Performance characteristics for in vitro diagnostic medical devices
- Technical documentation
- Meet harmonised standards and common specifications (An introductory guide to the medical device regulation (MDR ...)
Risk Classification
Technical File
At a minimum, technical documentation should have:
- A device description and specification section
- Labelling and instructions for use
- Detailed information on design and manufacturing
- Detailed risk management information in compliance with ISO 14971
- General Safety and Performance Requirements (GSPR) (formerly known as essential requirements)
- Verification and validation information. Not just design and development but also clinical evaluation (performance) to support device design and safety
- Post-market surveillance (PMS) information, including PMS plan, post-market clinical follow-up (PMCF) plan, and periodic safety update report (PSUR)
Clinical Evaluation
- The emphasis is to undertake clinical investigations to evaluate safety and performance of the device in accordance with the requirements of the regulatory authorities. Pre UKCA/CE approval studies may include:
- Evaluation of safety and performance against standard care
- Feasibility study to investigate device design and function against device specification
- To evaluate patient populations and clinical pathways
- Consider small studies with 10s or 100s of patients
- At this stage large scale randomised controlled trails (RCTs) are not normally undertaken due to time and cost
- Need a letter of no objection from regulatory body (MHRA) to use a non UKCA/CE marked device in a clinical investigation