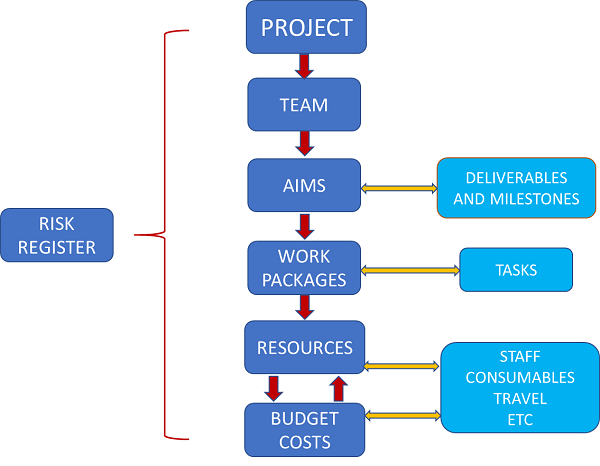
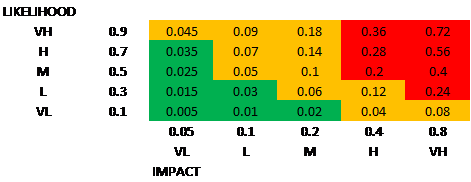
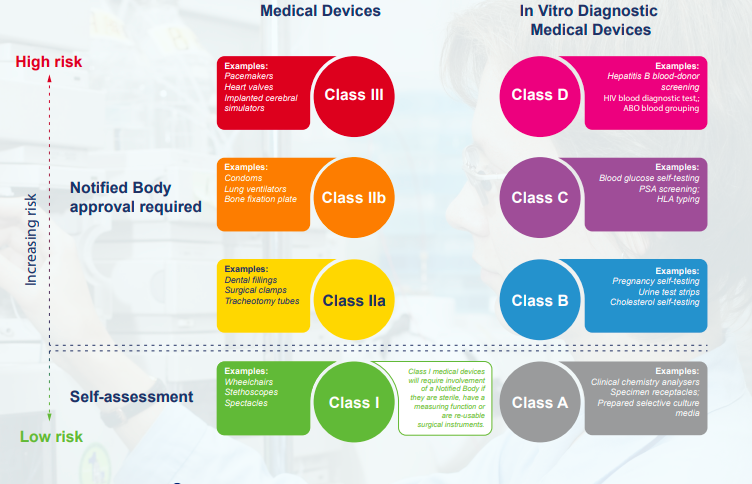
The first clinical study for a medical device should be aimed at supporting requirements for UKCA/CE marking. Prior evidence and any key data generated should be used to support these follow-on studies.
This study should ensure that safety and performance are clearly demonstrated (safety and acceptability/tolerability study; pilot or feasibility studies). A pilot study can be used to assess the feasibility of conducting a larger follow-on clinical trial. The pilot study will allow the determination of likely difficulties in performing a full clinical trial and inform on the calculations on sample sizes required for a full clinical trial to be done.
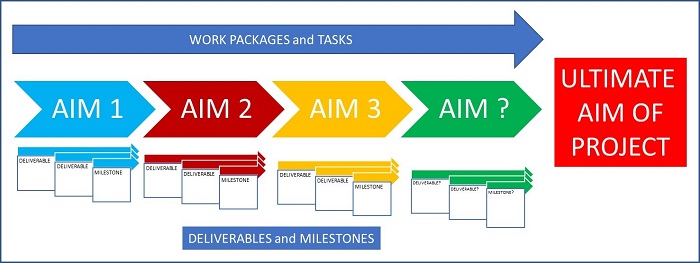
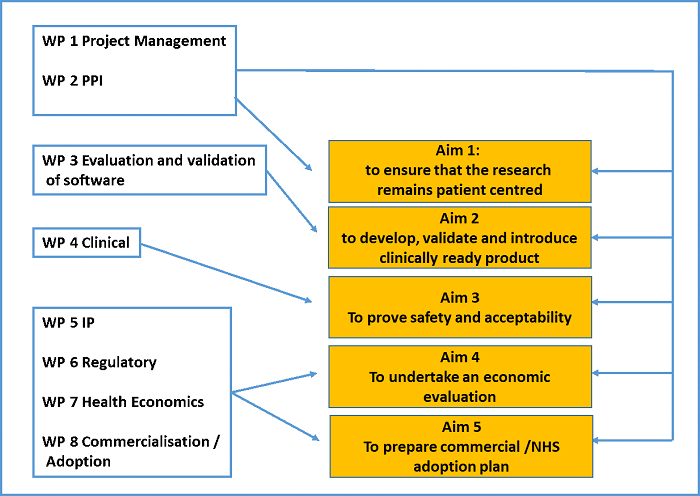
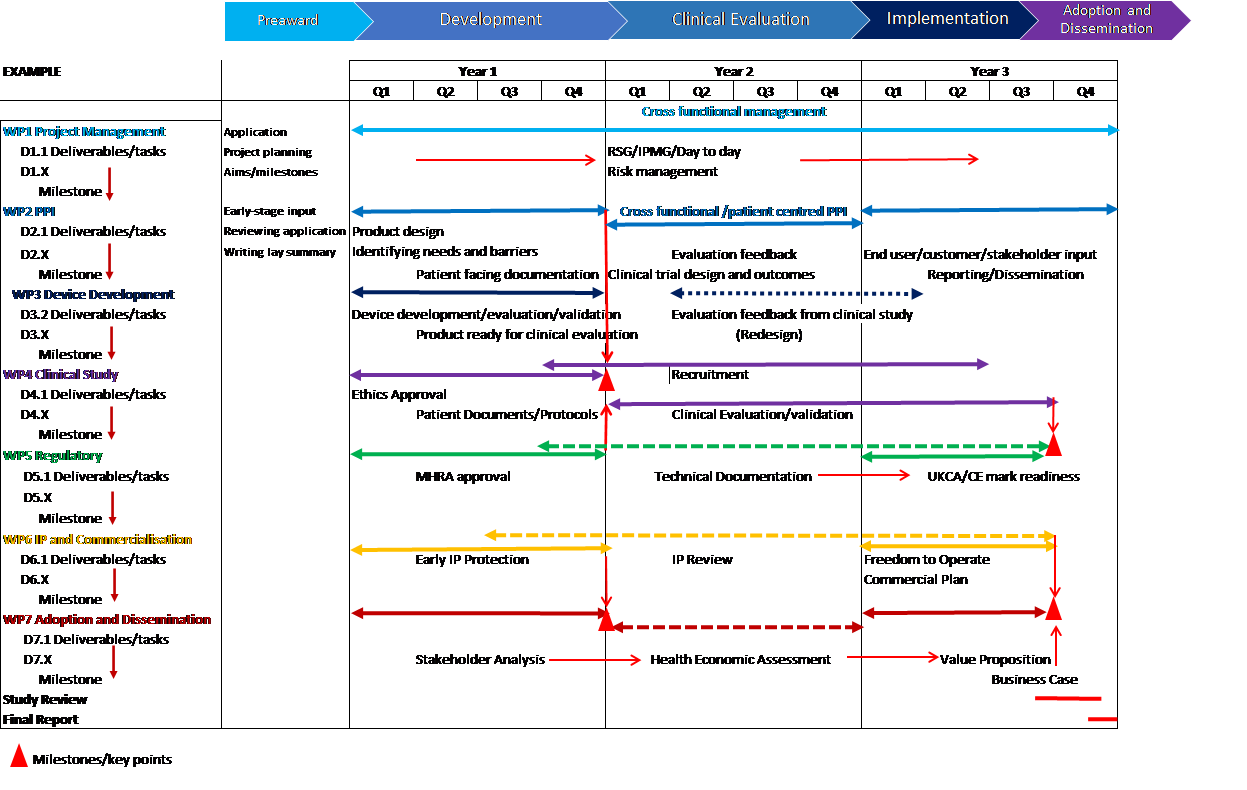
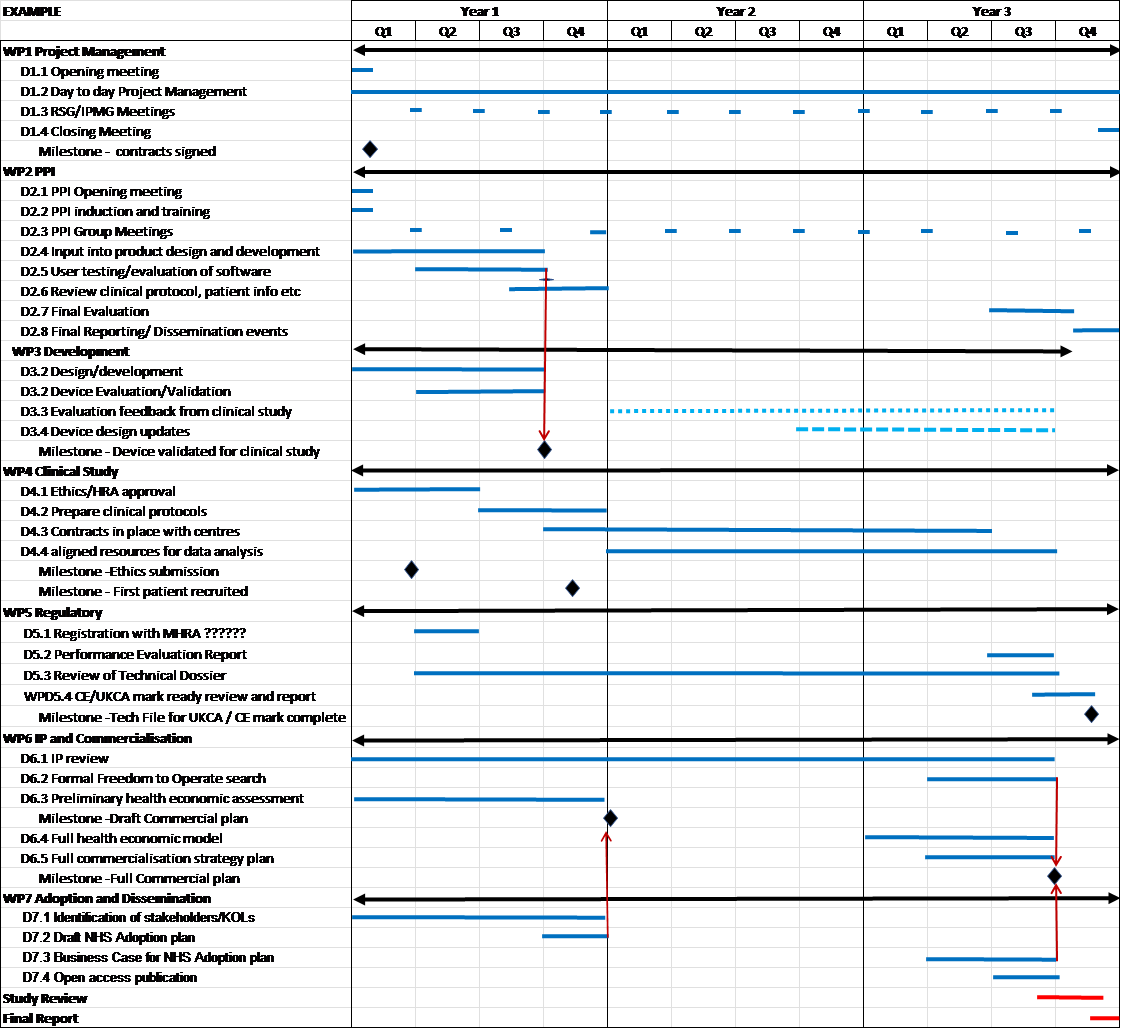
Thought should be given to how the clinical study supports the development of the device, how it could support economic evaluation and implementation into the clinical pathway.
Clinical Evaluation
For consideration:
- Emphasis should be to undertake clinical investigations to evaluate safety and performance of the device in accordance with the requirements of the regulatory authorities Pre UKCA/CE approval studies may include:
- Evaluation of safety and performance against standard care
- Feasibility study to investigate device design and function against device specification
- To evaluate patient populations and clinical pathways
- Consider small studies with 10s or 100s of patients
- At this stage, large-scale randomised controlled trials (RCTs) are not normally undertaken due to time and cost
- Need a letter of 'no objection' from the regulatory body (MHRA) to use a non UKCA/CE marked device in a clinical investigation
Regulatory/Ethics Approval
Ethical approval must be obtained either before or at the start of the project and should be part of study planning.
Before the clinical study commences an application should be made to the MHRA for non UKCA/CE marked device assessment.
Applicants can discuss ethical approval requirements with their Local Clinical Research Network (LCRN)and the Health Research Authority (HRA).
Applications for approval for a clinical investigation of a medical device should be made via the Integrated Research Application System (IRAS) website and by completing the Medical Devices form (Clinical Investigation application form). Upload the relevant supporting documents on to IRAS and then follow the instructions on how to submit the application.
The IRAS form also contains a checklist of documentation required for MHRA submission.
The Checklist tab on IRAS contains a list of all documents that should be included in the submission to MHRA:
- Covering letter on headed paper
- Clinical investigation plan
- Investigator’s brochure
- Participant information sheet
- Participant consent form
- CVs for UK clinical investigators
- Device details
- Essential requirements checklist/General Safety and Performance Requirements checklist
- Risk analysis
- Instructions for use of a medical device
- Device labels
- Summary of all bench testing and pre-clinical testing conducted
- Summary of all clinical experience with the device to date
- End-of-study reports for any concluded clinical investigations that involved the same
- Medical device under investigation
- List of standards met
- Sterilisation validation report (where relevant)
- Software information (where relevant)
- Biological safety assessments of patient contacting materials (where relevant)
- Information on animal tissues (where relevant)
- Information on any medicine or human blood derivative, or non-viable human tissues and cells incorporated into the device
- Research ethics committee opinion (if available)
Information on documentation required for submission can be found in the ‘Clinical investigations of medical devices – compiling a submission to MHRA’ document.
Some clinical evaluation parameters and design for consideration when putting protocol together:
- Aims and objectives of clinical study
- Type of investigation
- Sample size (with justification)
- Number of centres participating in the study with justification
- Duration of study with start and finish dates and proposed follow-up period (with justification)
- MHRA Guidance on legislation
- Criteria for patient selection including: Inclusion and exclusion criteria (with justification), criteria for withdrawal
- Details of any proposed post-market clinical follow-up
- Data collection/analysis/statistics
- Description of endpoints (primary and secondary) and the data recorded to achieve the endpoints, method of patient follow-up, assessment and monitoring
- The hypotheses which are to be tested and/or the device performance characteristics including statistical methods
- Description of procedures and details of data to record and report serious adverse events and adverse device-related incidents
Further information can be found in the ‘Clinical investigations of medical devices – compiling a submission to MHRA’ document.
Sponsorship for Clinical Studies
NIHR state that “if support for a clinical study is requested, one of the partners in the research project should be an NHS organisation which has agreed to be the sponsor of the trial”.
For clinical research, the sponsor will normally be the NHS Trust but it may also be a commercial company or a university. The sponsor can be the Principal Investigator's employing organisation.
Proof of an approved sponsor for all research taking place within NHS is required.
All research taking place in the NHS (where the research involves NHS patients, data or tissue) must have sponsor. The sponsor is the organisation that takes the lead and has primary responsibility for the design of the study meeting appropriate standards. The sponsor verifies that arrangements are in place to ensure the correct conduct for collecting and analysing of data and reporting.
Clinical Trials Units (CTU)
CTUs are specialist units which have been set up with a specific remit to design, conduct, analyse and publish clinical trials and other well-designed studies. They have the capability to provide specialist expert statistical, epidemiological and other methodological advice and coordination to undertake successful clinical trials. In addition, most CTUs will have expertise in the coordination of trials involving investigational medicinal products.
Having a CTU on board can be advantageous as it would demonstrate that they are committed to supporting the trial.
Governance
The UK Policy Framework for Health and Social Care Research outlines the principles of good practice in the management and conduct of health and social care research in the UK. All NIHR-funded research must be completed in accordance with this framework.
Patient Consent
People who are invited to take part in health and care research must give informed consent before being enrolled. For consent to be considered both legal and ethical it must be:
- Given by a person with capacity
- Voluntarily given, with no undue influence
- Given by someone who has been adequately informed
- A fair choice
Research funded or supported by the NIHR should follow best practice in consent and the preparation of information for participants, as set out by the Health Research Authority.
Protecting Personal Data
Researchers and study coordinators must comply with General Data Protection Regulation (GDPR) with respect to processing personal data, such as from research participants.
The Health Research Authority has published operational guidance on the implications of the GDPR for the delivery of research in the UK.